+1.310.385.1918 | 8436 W. 3rd St. #800 L.A.
Follow us:
![]()
![]()
![]()
![]()
![]()
+1.310.385.1918 | 8436 W. 3rd St. #800 L.A.
Follow us:
![]()
![]()
![]()
![]()
![]()

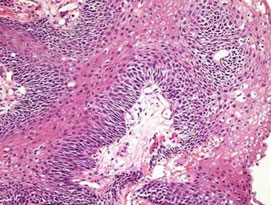
Craniopharyngiomas are epithelial tumors that arise from remnants of tissue present during the embryonic development of the foregut (Rathke’s pouch). Histologically similar to Rathke’s cleft cysts, they arise from or in the region of the pituitary stalk. As they grow they can compress or attach to the optic nerve, hypothalamus, frontal and temporal lobes of the brain as well as the adjacent blood vessels (eg internal carotid arteries) and cranial nerves. Tumor consistency is highly variable with some being primarily solid while others are cystic (fluid containing). Calcification and cholesterol crystal are commonly associated histologic features. Presenting symptoms include loss of vision, headache, personality changes, hormonal abnormalities, and developmental or growth delay in children.
A variety of treatment approaches have been developed. These include surgical excision, aspiration of the contents of the tumor, shunting procedures and various forms of radiation therapy. In many cases minimally invasive surgery through the nose (endonasal surgery) or eyebrow (supraorbital approach) have been very effective at extirpating craniopharyngiomas. In other cases surgery is limited by the tumors adherence to surrounding vital structures. The exact approach for surgery and treatment depends on the specific radiographic features, growth characteristics, tumor constency, and location in each individual patient.